The DMFT method is used to calculate the physical properties for the correlated systems. DMFT was originally developed [2] in the context of the Hubbard model considering the limit of large dimensions [12] (or connectivity), where DMFT becomes exact, but later it's managed to be consistent with Luttinger-Ward approach.
For more information upon the DMFT with new approach check the following link:
http://hauleweb.rutgers.edu/tutorials/whatis/whatis.html
The new version of the DMFT method as working within the Luttinger-Ward approach is implemented within the code called eDMFT.
To know how to use the DMFT method within the eDMFT code read the following picture
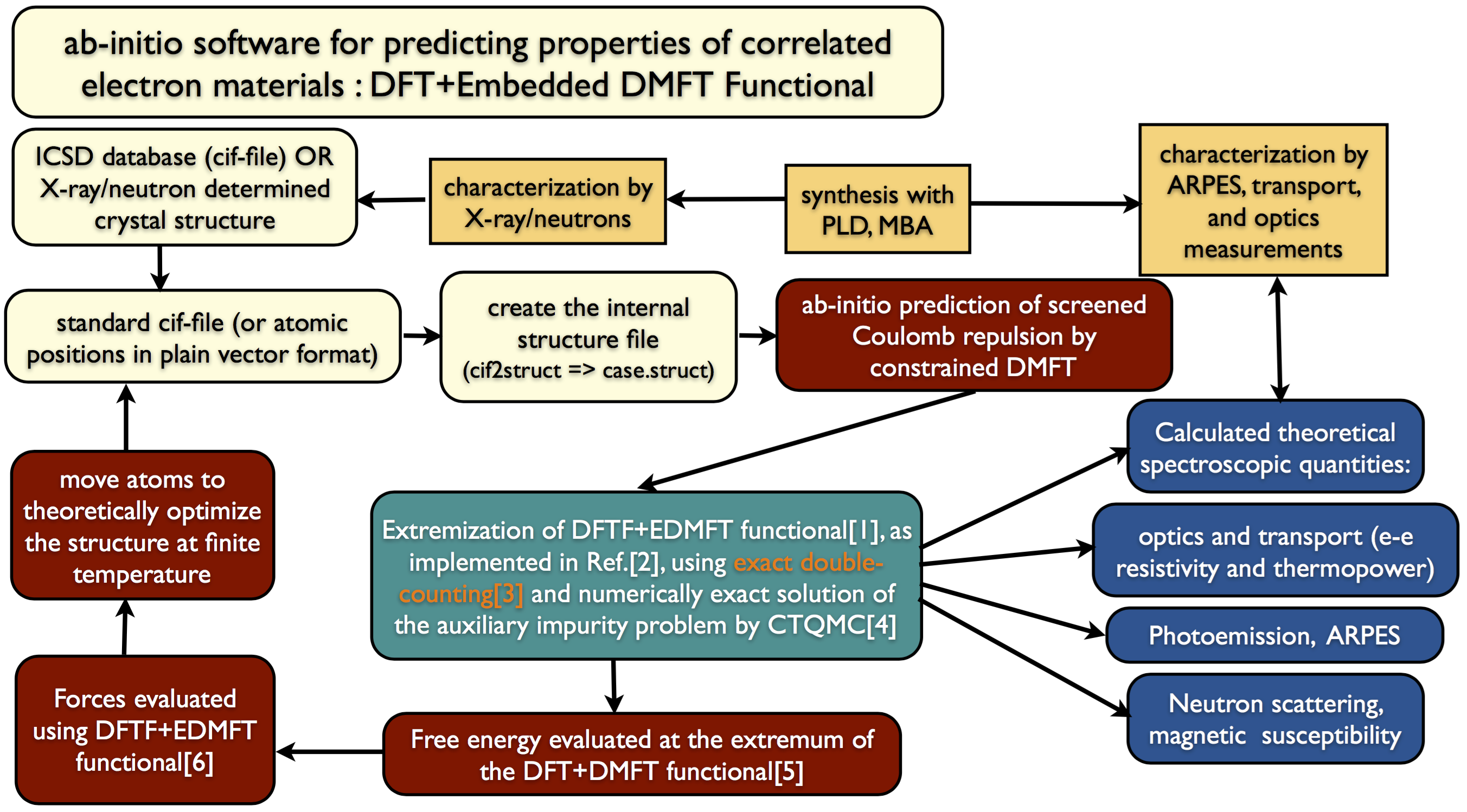
To know how does the DMFT method work with the wien2k code check the following pictures:
DFT within wien2k without DMFT
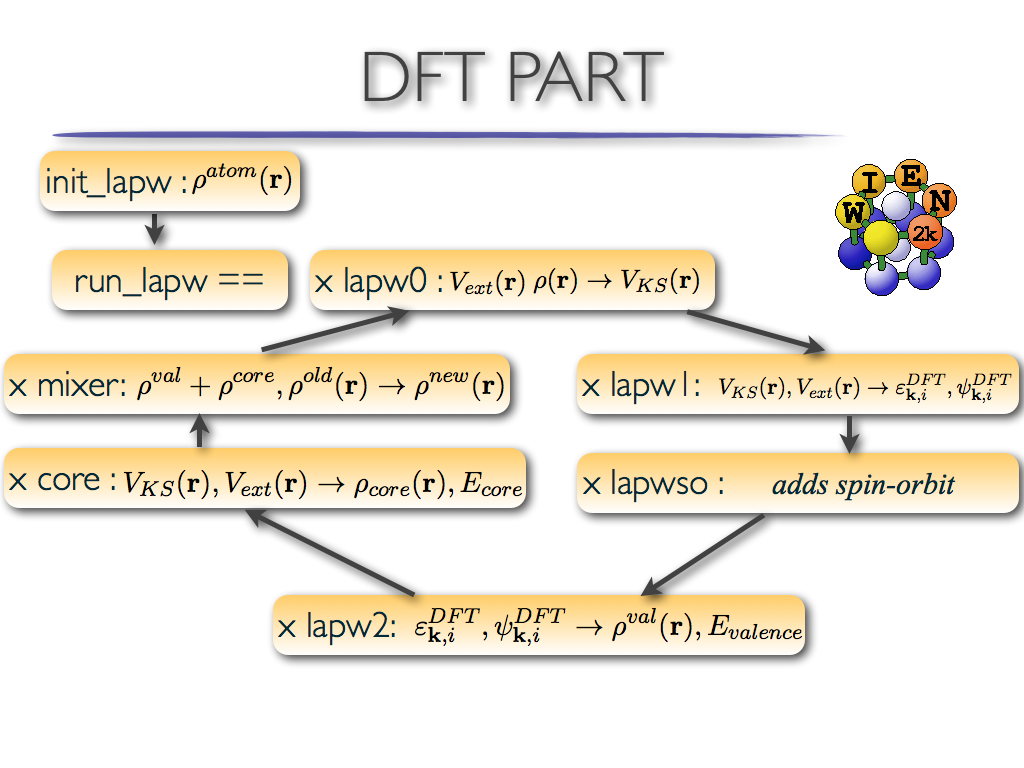
- lapw0: calculates electronic potential
- lapw1: solves eigenvalue problem for Kohn-Sham orbitals
- lapwso: adds spin-orbit coupling
- lapw2: calculates DFT valence electronic charge
- core: adds core states to DFT charge
- mixer: mixes electronic charge density
DFT within wien2k with DMFT method
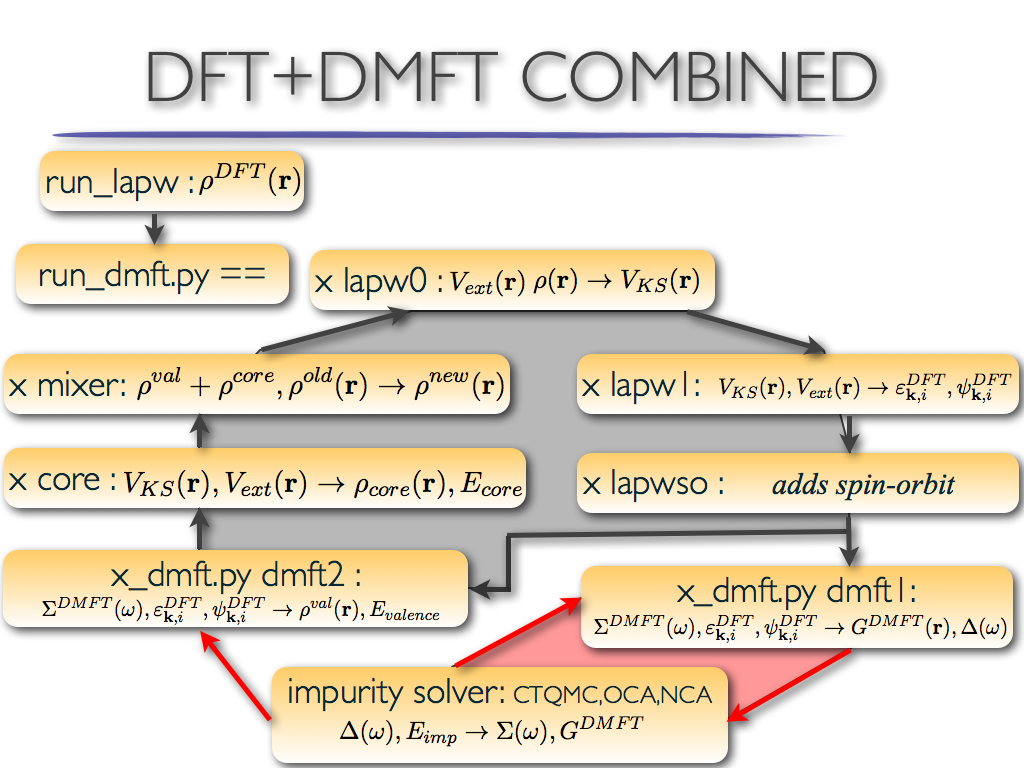
The eDMFT steps are:
- lapw0: calculates electronic potential
- lapw1: solves eigenvalue problem for Kohn-Sham orbitals
- lapwso: adds spin-orbit coupling
- dmft1: calculates local green's function and hybridization function
- impurity: solves the auxiliary impurity problem for electronic self-energy correction
- dmft2: calculates DMFT+DFT valence electronic charge
- core: adds core states to DFT charge
- mixer: mixes electronic charge density
For more information on how does the eDMFT code work check the following link:
Downloading the eDMFT code
The program consists of many independent programs and executables, which are written in C++, fortran90, and Python. Here you can download its
Installing the eDMFT code
To install the code you need to download some packages:
- C++ compiler (either gnu or intel)
- fortran compiler (either gnu or intel)
- Python (version 2) with numpy and scipy as well as weave (the latter became stand alone package, separate from scipy, after scipy 0.14 version)
- blas/lapack library (intel mkl or similar). If installation does not find it, it will try to download it and install it. You will be prompted.
- gsl library: gnu scienific library. If you do not have it, you will be prompted to do download and install it.
- fftw library for fast Fourier transform
- Finally, Wien2K needs to be installed to run charge self-consistent calculations.
The simplest type of compilation (after un-taring the code tar xzvf EDMFTF.tgz) consist of running the python setup script, i.e.,
To clean the previous compilation (all object files and libraries, but not removing the final binary files), you should type
python setup.py
To clean the previous compilation (all object files and libraries, but not removing the final binary files), you should type
python setup.py --clean
However, before you start compilation, it is advisable to first edit configure.py file, which contains information about existing libraries. There are several examples of the configuration script provided, for example, configure.py.macos, configure.py.intel, configure.py.gnu. If you are trying the code on MacOS, simply copy configure.py.macos to configure.py. On system with all intel compilers one can copy configure.py.intel to configure.py and then run python setup.py.
For more details check the following link:
After the installation is complete, the following variables should be set in your .bashrc (or equivalent):
WIEN_DMFT_ROOT PYTHONPATH WIENROOT SCRATCH EDITOR
The last three should be specified during Wien2k installation, and in general do not need to be changed now. It is however advisable to set SCRATCH to current directory. For example, typical installation will have the following variables set to
export WIEN_DMFT_ROOT=$HOME/EDMFT/bin export PYTHONPATH=$PYTHONPATH:$WIEN_DMFT_ROOT export WIENROOT=$HOME/wien2k export SCRATCH="." export EDITOR="emacs -nw" export PATH=$WIENROOT:$WIEN_DMFT_ROOT:$PATH
The properties calculated by the eDMFT code:
- total free energy and forces in the presence of strong correlations
- exact double-counting of DMFT and LDA
- non-collinear magnetism
- theoretical spectroscopies, such as optics, resistivity, neutron structure factor, resonant elastic X-ray scattering
- several solvers: continuous time quantum Monte Carlo, NCA, OCA
- extreme parallelism (scales up to 100,000 cores) using Message Passing Interface (MPI)
- parallel execution by OpenMP
To execute the eDMFT code there are many tutorials as follows:First Tutorial: Learn how to use the CTQMC impurity solver
Tutorial 6: Sr2IrO4, large spin-orbit and non-collinear magnetism









0 Comments