
For the treatment of the correlated electrons, there are 3 types of functionals used:
1- The on-site exact exchange functional
2- The on-site hybrid functioals
3- The full hybrid (screened and unscreened ) functionals
In WIEN2kit is also possible to go beyond standard LDA (GGA) and include on-site exact-exchange (Hartree-Fock), which is very usefull for strongly correlated systems. The exact-exchange/hybrid methods are implemented only inside the atomic spheres, therefore it is recommended to us them only for localized electrons (see Tran et al. 2006 for details). They will NOT improve gaps in sp-semiconductors.
A- On-site functionals:
For the on-site functionals (exact exchange and hybrid ), we need to use the 2 input files case.in0 and case.ineece.
The difference in the file case.ineece between on-site exact exchange and on-site hybrid functionals lie in the determination of the mode:
- On-site exact exchange functional : EECE HYBR / EECE mode
- On-site hybrid functional : HYBR HYBR / EECE mode
| HYBR means that LDA/GGA exchange will be replaced by the exact exchange |
| EECE means that LDA/GGA exchange-correlation will be replaced by the exact exchange |
Examples of implemented functionals:
- LDA-Hartree-Fock
Functional 5 in case.in0. mode = EECE and fraction = 1 in case.ineece.
![\begin{displaymath}
E_{xc}^{\textrm{LDA-HF}}[\rho] = E_{xc}^{\textrm{LDA}}[\rho]...
...{\textrm{corr}}] -
E_{xc}^{\textrm{LDA}}[\rho_{\textrm{corr}}]
\end{displaymath}](http://euler.phys.cmu.edu/cluster/WIEN2k/img124.png)
- LDA-Fock-
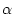
Functional 5 in case.in0. mode = HYBR and fraction = in case.ineece.
![\begin{displaymath}
E_{xc}^{{\textrm{LDA-Fock-}}\alpha}[\rho] = E_{xc}^{\textrm{...
...rm{corr}}] -
E_{x}^{\textrm{LDA}}[\rho_{\textrm{corr}}]\right)
\end{displaymath}](http://euler.phys.cmu.edu/cluster/WIEN2k/img125.png)
- PBE-Fock-
Functional 13 in case.in0. mode = HYBR and fraction = in case.ineece.
![\begin{displaymath}
E_{xc}^{\textrm{PBE-Fock-}\alpha}[\rho] = E_{xc}^{\textrm{PB...
...rm{corr}}] -
E_{x}^{\textrm{PBE}}[\rho_{\textrm{corr}}]\right)
\end{displaymath}](http://euler.phys.cmu.edu/cluster/WIEN2k/img126.png)
The PBE0 functional corresponds to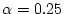 .
. - PBEsol-Fock-
Functional 19 in case.in0. mode = HYBR and fraction = in case.ineece.
- WC-Fock-
Functional 11 in case.in0. mode = HYBR and fraction = in case.ineece.
- TPSS-H-Fock-
Functional 27 in case.in0. mode = HYBR and fraction = in case.ineece.
It is similar to PBE0, but uses the meta-GGA TPSS. - B3PW91
Functional 18 in case.in0. mode = HYBR and fraction = 0.2 in case.ineece.
![\begin{eqnarray*}
E_{xc}^{\textrm{B3PW91}}[\rho] & = & E_{xc}^{\textrm{LDA}}[\rh...
...(E_{c}^{\textrm{PW91}}[\rho] -
E_{c}^{\textrm{LDA}}[\rho]\right)
\end{eqnarray*}](http://euler.phys.cmu.edu/cluster/WIEN2k/img131.png)
In addition to the input files which are necessary for an usual LDA or GGA calculation, the input file case.ineece is necessary to start a calculation. You may copy a template from SRC_templates, but must modify it according to your needs. In particular you must select for which atoms and which orbitals (usually d-Orbitals of late transition metal atoms or f-orbitals for 4f/5f atoms) you want to add such a potential and which type of functional you want to use.
A sample input for calculations with exact exchange is given below.
------------------ top of file: case.ineece ----------- -9.0 2 emin, natorb 1 1 2 1st atom index, nlorb, lorb 2 1 2 2nd atom index, nlorb, lorb HYBR HYBR / EECE mode 0.25 fraction of exact exchange ------------------ bottom of file ---------------------
As with LDA+, on-site functionals can be used only for spin-polarized calculations (runsp_lapw with the switch -eece). runsp_lapw will internally call runeece_lapw, which will create all necessary additional input files (it requires a case.in0 file including the optional IFFT line as generated by init_lapw): case.indm (case.indmc), case.inorb, case.in0eece, case.in2eece (case.in2ceece) and once this is done, calculates in a series of lapw2/lapwdm/lapw0/orb calculations the corresponding orbital dependend potentials.
- runsp_lapw -eece [-so]
B- Full hybrid functionals:
The full hybrid (screened and unscreened ) need to use the 3 input files case.in0 , case.in0_grr and case.inhf.
The onsite exact-exchange/hybrid functionals can be applied only to localized electrons (typically 3d or 4f), but lead to cheap calculations. In WIEN2k, it is also possible to apply hybrid (and Hartree-Fock) functionals to all electrons, however this leads to calculations which are one or two orders of magnitude more expensive. Hybrid functionals are usually more accurate than the semilocal functionals for the electronic properties of semiconductors and insulators. They also give accurate results for strongly correlated systems like NiO. In hybrid functionals a fraction α of semilocal (SL) exchange is replaced by Hartree-Fock (HF) exchange:
E hybrid xc = E SL xc + α E HF x − E SL x .
Hybrid functionals can also be constructed by considering only the short-range part of EHF x and ESL x , which leads to the so-called ”screened” hybrid functionals. In WIEN2k, the unscreened and screened hybrid functionals are implemented using the second-variational procedure (Tran and Blaha 2011).
The available functionals
Among the semilocal functionals ESL xc available in WIEN2k, only a few of them can be used in Ehybrid xc (both in unscreened and screened modes). The functionals are the following (case.in0 grr is for the exchange part ESL x ):
LDA: XC LDA (indxc=5) in case.in0 and EX SLDA VX SLDA (indxc=51) in case.in0 grr I
PBE: XC PBE (indxc=13) in case.in0 and EX SPBE VX SPBE (indxc=52) in case.in0 grr.
The functionals PBE0 and YS-PBE0 (similar to HSE06) correspond to α = 0.25 in case.inhf (see 7.4).
WC: XC WC (indxc=11) in case.in0 and EX S WC VX SWC (indxc=53) in case.in0 grr I PBEsol: XC
PBESOL (indxc=19) in case.in0 and EX SPBESOL VX SPBESOL (indxc=54) in case.in0 grr
BPW91: EX B88 VX B88 EC PW91 VC PW91 (indxc=17) in case.in0 and EX SB88 VX SB88 (indxc=55) in case.in0 grr. (this is not B3PW91).
BLYP: EX B88 VX B88 EC LYP VC LYP (indxc=24) in case.in0 and EX SB88 VX SB88 (indxc=55) in case.in0 grr. (this is not B3LYP).
The onsite exact-exchange/hybrid functionals can be applied only to localized electrons (typically 3d or 4f), but lead to cheap calculations. In WIEN2k, it is also possible to apply hybrid (and Hartree-Fock) functionals to all electrons, however this leads to calculations which are one or two orders of magnitude more expensive. Hybrid functionals are usually more accurate than the semilocal functionals for the electronic properties of semiconductors and insulators. They also give accurate results for strongly correlated systems like NiO. In hybrid functionals a fraction α of semilocal (SL) exchange is replaced by Hartree-Fock (HF) exchange:
E hybrid xc = E SL xc + α E HF x − E SL x .
Hybrid functionals can also be constructed by considering only the short-range part of EHF x and ESL x , which leads to the so-called ”screened” hybrid functionals. In WIEN2k, the unscreened and screened hybrid functionals are implemented using the second-variational procedure (Tran and Blaha 2011).
The available functionals
Among the semilocal functionals ESL xc available in WIEN2k, only a few of them can be used in Ehybrid xc (both in unscreened and screened modes). The functionals are the following (case.in0 grr is for the exchange part ESL x ):
LDA: XC LDA (indxc=5) in case.in0 and EX SLDA VX SLDA (indxc=51) in case.in0 grr I
PBE: XC PBE (indxc=13) in case.in0 and EX SPBE VX SPBE (indxc=52) in case.in0 grr.
The functionals PBE0 and YS-PBE0 (similar to HSE06) correspond to α = 0.25 in case.inhf (see 7.4).
WC: XC WC (indxc=11) in case.in0 and EX S WC VX SWC (indxc=53) in case.in0 grr I PBEsol: XC
PBESOL (indxc=19) in case.in0 and EX SPBESOL VX SPBESOL (indxc=54) in case.in0 grr
BPW91: EX B88 VX B88 EC PW91 VC PW91 (indxc=17) in case.in0 and EX SB88 VX SB88 (indxc=55) in case.in0 grr. (this is not B3PW91).
BLYP: EX B88 VX B88 EC LYP VC LYP (indxc=24) in case.in0 and EX SB88 VX SB88 (indxc=55) in case.in0 grr. (this is not B3LYP).
In addition, calculations with the well-known B3PW91 and B3LYP (with VWN5) unscreened hybrid
functionals (see 4.5.7 for the functionals form) can also be done:
B3PW91: XC B3PW91 (indxc=18) in case.in0, EX SLDA VX SLDA (indxc=51) in case.in0 grr and α = 0.2 in case.inhf
B3LYP: XC B3LYP (indxc=47) in case.in0, EX SLDA VX SLDA (indxc=51) in case.in0 grr and α = 0.2 in case.inhf
Hartree-Fock calculations (without correlation) are also possible:
HF: EX LDA VX LDA (indxc=6) in case.in0, EX SLDA VX SLDA (indxc=51) in case.in0 grr and α = 1 in case.inhf
How to do a calculation with full hybrid functional
Flow in run lapw -hf
The flow of programs during a scf iteration when executing run lapw -hf is the following (nonspin-polarized and real case):
B3PW91: XC B3PW91 (indxc=18) in case.in0, EX SLDA VX SLDA (indxc=51) in case.in0 grr and α = 0.2 in case.inhf
B3LYP: XC B3LYP (indxc=47) in case.in0, EX SLDA VX SLDA (indxc=51) in case.in0 grr and α = 0.2 in case.inhf
Hartree-Fock calculations (without correlation) are also possible:
HF: EX LDA VX LDA (indxc=6) in case.in0, EX SLDA VX SLDA (indxc=51) in case.in0 grr and α = 1 in case.inhf
How to do a calculation with full hybrid functional
Flow in run lapw -hf
The flow of programs during a scf iteration when executing run lapw -hf is the following (nonspin-polarized and real case):
- x lapw0 -grr (semilocal exchange)
- x lapw0 (semilocal exchange-correlation)
- x lapw1 (semilocal orbitals)
- x lapw2 (semilocal bands)
- mv case.vectorhf case.vectorhf old
- x hf (hybrid orbitals)
- cp case.klist fbz case.klist, cp case.kgen fbz case.kgen
- x lapw2 -hf (hybrid electron density and bands)
- cp case.klist ibz case.klist, cp case.kgen ibz case.kgen
- x lcore
- x mixer
Self-consistent calculation
The steps to perform a calculation with hybrid functionals are the following:
- Do a calculation with the underlying semilocal functional ESL xc (recommended but not mandatory).
- ”save” the semilocal calculation.
The next steps can be done conveniently using init_hf_lapw [-up]:
- I create case.inhf (cp $WIENROOT/SRC templates/template.inhf case.inhf). While there are (more or less) reasonable default values for most parameters, you must set nband manually. Set nband to at least the number of occupied bands plus one. Larger values are more accurate, but be aware that computing time scales as nband2 . The value of nband which leads to a converged result will strongly depend on the studied system and property (e.g., much higher for EFG than for band gap or lattice constant).
- I create case.in0 grr (cp case.in0 case.in0 grr), this file contains: – a screened exhccange functional (EX SLDA, ... or indxc=51, 52, 53, 54 or 55) for the exchange functional (see above) – ”R2V” option (instead of ”NR2V”) such that the exchange potential is written to case.r2v grr – ”KXC” (instead of ”TOT”) such that αESL x is printed in case.scf0 grr (and case.scf) under the label :AEXSL
- IIn case.inc the print switch has to be ”1” for all atoms such that the core orbitals are printed in case.corewf (you don’t have to set this manually, the script run lapw will do it automatically when -hf is specified).
- I if nband is large, you may have to edit case.in1 and set EMAX to a higher value (e.g., 5 Ry).
- I Execute run kgenhf lapw. This generates case.klist fbz, case.kgen fbz, case.klist ibz, case.kgen ibz and case.outputkgenhf. One must use identical k-meshes and shifts for IBZ and FBZ. Note that the k-parallelization is done over the k-points specified in case.klist ibz (irreducible Brillouin zone).
Once the initializations has been done, execute run lapw (or runsp lapw) with the switch -hf: run(sp) lapw -hf ...
Neglect of the nondiagonal terms
If only the eigenvalues are wanted, you may use the switch -diaghf. By using this switch, only the diagonal elements of the 2nd variational Hamiltonian matrix are calculated (the non-diagonal elements are set to zero). This leads to a much faster calculation of the eigenvalues, while keeping a very good accuracy (Tran 2012). However, the orbitals will not be modified, therefore running the calculation for more than one iteration is useless (the result will not change except for metallic systems). This option is not recommended for systems which are described as metallic with the semilocal functional or for difficult systems (e.g., NiO, see Tran 2012). It is important to be aware that with this option, the total energy (:ENE in case.scf) is nonsense unless the option -nonself (see below) was also used. After having done and saved a well converged calculation with the semilocal functional, the setting up of such a calculation is the same as for a self-consistent calculation (see above), but then run(sp) lapw is executed with -diaghf (-hf and -i 1 will be set automatically):
run(sp) lapw -diaghf ...
In order to reduce the computational time for the calculation of the HF potential, the internal loop
over the k-points can be reduced to a subset of k-points. For instance, for a calculation with a
12×12×12 k-mesh, the reduced k-mesh for the HF potential can be one of the following: 6×6×6,
4 × 4 × 4, 3 × 3 × 3, 2 × 2 × 2 or 1 × 1 × 1. This option, which should be used only in the case of
screened exchange (see Paier et al. 2006), is particularly interesting for total energy calculations.
Obviously, choosing such a reduced k-mesh is an approximation which needs to be tested. The
setting up of such a calculation is the same as for a self-consistent calculation (see above), but with
the switch -redklist when executing run kgenhf lapw (to create also case.klist rfbz):
run kgenhf lapw -redklist
and run(sp) lapw: run(sp) lapw -hf -redklist ...
Non-self-consistent calculation of the total energy for hybrid functionals
It is possible to calculate the total energy non-self-consistently, i.e., by plugging the orbitals obtained
from a calculation with the underlying semilocal functional ESL
xc into the total-energy hybrid
functional. By doing so, the 2nd variational Hamiltonian is not calculated and therefore the
computational time will be reduced. After having done and saved a well converged calculation
with the semilocal functional, the setting up of a non-self-consistent calculation is the same as for
a self-consistent calculation (see above), but with the additional switch -nonself (-hf and -i 1
will be set automatically) that has to be used in run(sp) lapw :
run(sp) lapw -nonself ...
This option can be particularly interesting for the calculation of the lattice constant, which depends
mainly on the functional, but very little on the orbitals plugged into the functional. It can be used
simultaneously with the option -diaghf (see above).
Starting a calculation from another k-mesh
Due to the orbital-dependence of the HF potential, it is not straightforward to start directly a calculation
with a potential generated from a previous calculation with another k-mesh. However, due
to the high cost of a hybrid calculation, it is desirable to have this possibility in order to reduce the
number of iterations during the scf procedure.
This option is also useful if a vector file on a very dense k-mesh is needed, e.g. for optics or
transport properties (BoltzTraP), while using such a k-mesh for a full self-consistent calculation
would not be necessary (and would be too expensive). In this case you want to do only one iteration
(-i 1).
The procedure is the following:
- Do the calculation with the first k-mesh and ”save” it when it is finished (do not execute clean lapw since case.vectorhf should be present).
- cp case.klist fbz case.klist fbz old
- cp case.klist rfbz case.klist rfbz old (if the option -redklist is also used) I Execute run kgenhf lapw to create the files for the new k-mesh.
- Run the HF calculation with -newklist: run(sp) lapw -hf -newklist (-i 1) ...
This option can be used simultaneously with -redklist but not with -diaghf (see above).
Band structure plotting
In order to make a plot of the band structure with hybrid functionals, it is more convenient to use
the program run bandplothf lapw. After the self-consistent calculation is finished and saved
(do not execute clean lapw since case.vectorhf must be present), do the following steps:
- Create case.klist_band.
- Execute run bandplothf lapw with one or several of the following flags that were also used during the self-consistent calculation: -up/dn, -diaghf, -redklist and -in1orig. Note that a parallel calculation of the band structure (with -p) can be done even if the scf calculation was not done in parallel (but you still need a file .machines). You can also use -qtl to calculate the partial charges for band character plotting.
- Create case.insp.
- Execute x spaghetti with the switch -hf.
First, run bandplothf lapw calculates the semilocal orbitals (x lapw1 -band) for the k-points
in case.klist band. Then, the hybrid eigenvalues at these k-points are calculated (x hf
-band). If -qtl is used, then the partial charges will be calculated (x lapw2 -band -qtl).
Density of states
The calculation of the DOS is the same as for the semilocal functionals, but using the additional flag
-hf when executing lapw2 for the partial charges (x lapw2 -qtl -hf) and tetra for the DOS (x
tetra -hf).








{kind=link}
1 Comments
We have sell some products of different custom boxes.it is very useful and very low price please visits this site thanks and please share this post with your friends. Warmtepomp
ReplyDelete